
Co to jest MLD?
Leukodystrofia metachromatyczna (ang. metachromatic leukodystrophy, MLD) to ultrarzadka, zagrażająca życiu, dziedziczna choroba neurometaboliczna. Pierwszy opis leukodystrofii metachromatycznej pojawił się w literaturze medycznej w 1925 roku.
MLD wywołuje mutacja w genie arylosulfatazy A (ARSA), która powoduje gromadzenie się materiałów zwanych sulfatydami w różnych częściach ciała, w tym w mózgu, nerwach obwodowych, a także w narządach takich jak wątroba, pęcherzyk żółciowy i nerki. Gromadzenie się sulfatydów powoduje uszkodzenie osłonki mielinowej – ochronnej warstwy otaczającej nerwy. Zniszczenie mieliny zakłóca prawidłowe funkcjonowanie układu nerwowego, co prowadzi do poważnych problemów neurologicznych, takich jak utrata zdolności ruchowych, trudności z mówieniem, a w zaawansowanych stadiach choroby – utrata podstawowych funkcji życiowych.
MLD jest lizosomalną chorobą spichrzeniową, dziedziczoną w sposób autosomalny recesywny. Oznacza to, że dziecko musi odziedziczyć wadliwy gen od obojga rodziców. MLD dotyka zarówno chłopców, jak i dziewczynki.
Osoby cierpiące na leukodystrofię metachromatyczną (MLD) mogą doświadczać różnorodnych problemów, takich jak trudności z mobilnością, zachowaniem i nauką. W miarę postępu choroby pojawiają się coraz większe trudności z poruszaniem się, mówieniem, połykaniem, jedzeniem oraz widzeniem.
Objawy, wiek ich wystąpienia oraz tempo postępu choroby różnią się w zależności od postaci MLD i obszarów mózgu, które są dotknięte. Najczęstszą formą jest postać o wczesnym początku – ponad połowa osób z MLD zaczyna wykazywać objawy przed ukończeniem 3. roku życia.
Częstość występowania leukodystrofii metachromatycznej w Europie wynosi 1/40 000 – 1/100 000. W Polsce nie ma rejestru pacjentów z MLD, w związku z tym nie ma danych epidemiologicznych dotyczących naszego kraju. Jeśli przyjmiemy, że choroba występuje z częstością 1 przypadka na 40 tys., oznacza to, że rocznie na świat przychodzi w Polsce kilkoro dzieci z MLD.
Objawy MLD
Początkowo choroba jest bezobjawowa. Dziecko zwykle prawidłowo się rozwija, nie dając rodzicom powodów do niepokoju. Pierwsze objawy nieleczonego, powoli rozwijającego się MLD pojawiają się najczęściej między 6. a 30. miesiącem życia, w postaci trudności w chodzeniu, drżeń, osłabienia siły mięśniowej i problemów z równowagą (fenotyp ruchowy). Objawy te są zwykle nieoczywiste i łatwe do pomylenia z innymi, mniej niebezpiecznymi zaburzeniami.
Do pierwszych wyraźnych objawów należy zatrzymany lub opóźniony chód czy nagłe, pozornie nieuzasadnione upadki. W dalszej kolejności dochodzi do stopniowej utraty uzyskanych wcześniej kamieni milowych rozwoju, w tym możliwości samodzielnego pozycjonowania ciała, mowy, gryzienia, przełykania itp.
U starszych dzieci, nastolatków lub dorosłych w pierwszej kolejności można dostrzec deficyty i regresję związane z funkcjami poznawczymi czy zmianami w zachowaniu. W kolejnym etapie objawy nasilają się aż dochodzi do tzw. fazy gwałtownej progresji, skutkującej niepełnosprawnością neurologiczną obejmującą zarówno niepełnosprawność fizyczną, jak i intelektualną.
Objawy MLD mogą zatem obejmować: opóźnienia rozwojowe u dzieci, trudności w chodzeniu, postępującą utratę umiejętności, zmiany w zachowaniu, niepełnosprawność intelektualną, spastyczność mięśniową, ból, drgawki.
Pediatra lub lekarz rodzinny często jako pierwszy obserwuje u dziecka nieprawidłowości rozwoju psychoruchowego. Pacjenta z podejrzeniem MLD należy pilnie skierować na diagnostykę w warunkach oddziału neurologii dziecięcej lub chorób metabolicznych.
PRZEBIEG KLINICZNY MLD
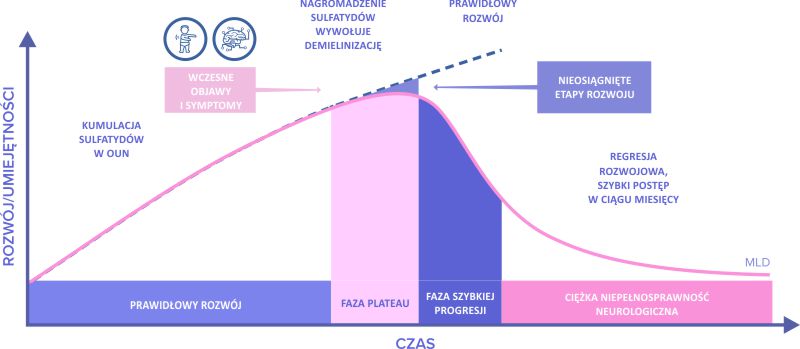
von Figura K et al. Metachromatic leukodystrophy 2001 In: The metabolic and molecular bases of inherited disease, Vol. 3, 8th ed. New York, NY: McGraw-Hill, 2001:3695-372. 2. Rosenberg JB et al. J Neurosci Res 2016;94(11):1169-79.
Kliniczne podtypy MLD
Objawy, wiek ich wystąpienia oraz tempo postępu choroby różnią się w zależności od postaci leukodystrofii metachromatycznej (MLD) i obszarów mózgu, które są dotknięte.
Choć wynikają z tej samej choroby, w zależności od etapu rozwojowego, mogą mieć różne nasilenie i charakter. Rokowanie i tempo rozwoju choroby również zależy od tego, w jakim momencie życia pojawią się jej pierwsze objawy. Im młodszy pacjent, tym szybszy spodziewany postęp choroby i krótsza oczekiwana długość życia. Jej przebieg jest wolniejszy u nastolatków i dorosłych.
Wyodrębniono cztery kliniczne podtypy MLD:
- późny niemowlęcy ≤30 miesiąca życia
ang.late infantile MLD / LI-MLD - wczesny młodzieńczy 2,5 – 6 rok życia
ang.early juvenile MLD / EJ-MLD - późny młodzieńczy 6 – 16 rok życia
ang. late juvenile MLD / LJ-MLD - dorosły ˃16 roku życia
ang. adult MLD
Odmiana późnoniemowlęca
LI-MLD
Najpowszechniejsza z odmian. Szacuje się, że dotyczy ponad połowy wszystkich zachorowań
Dotyczy 50%-60% pacjentów z MLD
Pierwsze objawy występują zwykle między 6 a 30 miesiącem życia po okresie pozornie prawidłowego rozwoju. Charakteryzuje je opóźnione osiąganie kamieni milowych w rozwoju dziecka – początkowe zahamowanie, a następnie regres – utrata uzyskanych wcześniej przez dziecko umiejętności motorycznych, spowolnienie i zaburzenie chodu.
U pacjenta zaobserwować można powrót takich refleksów neurologicznych jak objaw Babińskiego czy odruch Moro. Rozwój choroby prowadzi do zaburzenia mowy, przełykania, wzroku (zanik nerwu wzrokowego, zez, oczopląs) i słuchu, zaburzenia chodu (możliwe dolegliwości bólowe kończyn, hipotonia mięśniowa przechodząca w spastyczność), neuropatii obwodowej, dystonii, stopniowego paraliżu, ataksji, dyzartii, objawów opuszkowych lub rzekomoopuszkowych. Pacjenci często doświadczają epilepsji. Mediana zgonu 4,2 lata.
Odmiana młodzieńcza
J-MLD
Obejmuje postać wczesną młodzieńczą (EJ-MLD) oraz późną młodzieńczą (LI-MLD)
Dotyczy 20%-30% pacjentów z MLD
Początek objawów występuje między 30. miesiącem życia, a 16 rokiem życia z zachowaną kolejnością pojawiania się objawów. Objawy są podobne do postaci późnoniemowlęcej, ale postępują wolniej. Do pierwszych objawów postaci młodzieńczej zalicza się zwykle problemy behawioralne oraz trudności w nauce (zaburzenia zachowania, zachowania impulsywne, zaburzenia snu, zaburzenia poznawcze, w tym problemy z koncentracją uwagi).
Stopniowo pojawiają się jednak zaburzenia motoryki dużej i małej. Zaburzenia motoryki występujące jako pierwsze objawy są niekorzystnym czynnikiem prognostycznym. Ból wynikający z neuropatii może być początkowo mylony z bólami wzrostowymi. Pacjent z czasem mierzy się ze spastyką rąk i nóg, pogorszeniem i utratą mowy, zaburzeniami widzenia, rozwijającą się dysfagią, epilepsją, ataksją.
Chory stopniowo traci funkcje kognitywne, występują objawy psychiatryczne. W przypadku tej postaci pacjent dożyć może swojej drugiej, a nawet trzeciej dekady życia. Mediana zgonu 17.4 lat.
Odmiana dorosła
adult MLD
Dotyczy 15%-20% pacjentów z MLD
Diagnozowana jest u pacjentów powyżej 16. roku życia, ale objawy mogą nie wystąpić nawet do 4 dekady życia. Podobnie jak w przypadku młodzieńczej MLD (J-MLD), zwykle do pierwszych symptomów należą zaburzenia behawioralne i emocjonalne oraz większa skłonność do uzależnień (narastające deficyty intelektualne, psychozy i inne zaburzenia psychiatryczne, zaburzenia zachowania, zmienność emocjonalna). W wyniku tego niejednokrotnie początkowo chorzy diagnozowani mogą być w kierunku zaburzeń psychicznych. Objawy rozwijają się zdecydowanie wolniej niż u młodszych pacjentów. Podobnie postęp choroby. Do rozwijających się zaburzeń kognitywnych dochodzą stopniowo objawy motoryczne (ataksja, łagodna neuropatia obwodowa lub jej brak) oraz padaczka.
Częstość występowania napadów padaczkowych w MLD waha się od 14 do 39%. Napady padaczkowe są częstsze w późniejszych stadiach MLD i zwykle dobrze odpowiadają na leczenie przeciwpadaczkowe.
Oprócz objawów neurologicznych kumulacja sulfatydów może również powodować manifestację trzewną. Najczęściej obserwuje się zajęcie pęcherzyka żółciowego w postaci pogrubienia ściany pęcherzyka żółciowego czy zapalenia pęcherzyka żółciowego.
SŁOWNICZEK POJĘĆ
ataksja
niezborność ruchów
demielinizacja
rozpad osłonek mielinowych w ośrodkowym lub obwodowym układzie nerwowym
dysfagia
zaburzenia połykania
dystonia
niekontrolowane, powtarzające się ruchy mięśni
dyzartria
zaburzenie mowy
epilepsja
padaczka
hipotonia mięśniowa
obniżone napięcie mięśniowe
manifestacja trzewna
w medycynie odnosi się do objawów lub symptomów związanych z narządami wewnętrznymi (tzw. trzewnymi)
MRI mózgu
rezonans magnetyczny mózgu
neuropatia obwodowa
uszkodzenie nerwów obwodowych, które może objawiać się mrowieniem, zaburzeniami równowagi, drżeniem i skurczami mięśni
objaw Babińskiego
odruchowe wyprostowanie palucha z jego zgięciem grzbietowym w trakcie drażnienia skóry boczno-dolnej powierzchni stopy
objawy opuszkowe i rzekomoopuszkowe
problemy z połykaniem oraz zaburzenia mowy
odruch Moro
odruch obejmowania, odruch występujący w odpowiedzi na gwałtowną zmianę położenia ciała noworodka, nagły hałas, ostry dźwięk
OUN
ośrodkowy układ nerwowy: mózg i rdzeń kręgowy
skala GMFC-MLD
ang. Gross Motor Function Classification in MLD, klasyfikacja funkcji motoryki dużej
spastyka
nadmierne napięcie mięśniowe
Jak wykrywa się MLD?
Testem ostatecznie potwierdzającym MLD jest badanie laboratoryjne aktywności enzymu ARSA i pomiar poziomu sulfatydów oraz badanie genetyczne, które stwierdza występowanie nieprawidłowości w obrębie genu ARSA.
Na etapie diagnostyki może być wykonane również badanie MRI mózgu, w którym lekarz może stwierdzić występowanie zmian metachromatycznych w obrębie istoty białej. Należy jednak pamiętać, że to badanie nie wykaże choroby przed wystąpieniem spowodowanych nią zmian w mózgu – oznacza to, że prawidłowy wynik wykonanego badania nie może stanowić podstawy do wykluczenia choroby.
Brak korelacji między obrazem klinicznym MLD a obrazem mózgu w MRI wynika z faktu, że demielinizacja obwodowego układu nerwowego często wyprzedza w czasie demielinizację OUN i pojawienie się charakterystycznych zmian w obrazowaniu mózgu.
W przypadku badań przesiewowych noworodków ocenia się poziom sulfatydów i enzymu ARSA badanie enzymatyczne i dokonuje potwierdzenia genetycznego choroby.
ALGORYTM DIAGNOSTYCZNY MLD
SKRINING SELEKTYWNY
kliniczne podejrzenie MLD
SKRINING PRZEDOBJAWOWY
badanie przesiewowe noworodków, skrining rodzinny
ANALIZA STĘŻENIA SULFATYDÓW
W SUCHEJ KROPLI KRWI
JEŚLI PODWYŻSZONE STĘŻENIE SULFATYDÓW
ANALIZA AKTYWNOŚCI ARYLOSULFATAZY A (ARSA)
w SUCHEJ KROPLI KRWI
JEŚLI OBNIŻONA AKTYWNOŚĆ ARSA
ANALIZA MOLEKULARNA
GENY ARSA, PSAP, SUMF1
Teresa H.Y. WU Improving newborn screening test performance for metachromatic leukodystrophy: Recommendation from a pre-pilot study that identified a late-infantile case for treatment.
Lucia Laugwitz,Newborn screening in metachromatic leukodystrophy – European consensus-based recommendations on clinical management.
Diagnostyka wrodzonych chorób metabolicznych, w tym leukodystrofii metachromatycznej, opiera się na zasadzie skriningu selektywnego.
Badanie w kierunku MLD należy rozważyć w każdym wieku, zwłaszcza w przypadku neuropatii obwodowej, opóźnienia lub regresu rozwoju psychoruchowego, demielinizacji w obrazowaniu mózgu czy anomalii pęcherzyka żółciowego.
Dlaczego moje dziecko zachorowało?
Leukodystrofia metachromatyczna jest dziedziczona autosomalnie recesywnie, co oznacza, że oboje rodzice muszą być bezobjawowymi nosicielami mutacji genetycznej (klinicznie zdrowi). W momencie poczęcia każde rodzeństwo osoby chorej ma 25% prawdopodobieństwa zachorowania, 50% prawdopodobieństwa bezobjawowego nosicielstwa i 25% prawdopodobieństwa, że nie będzie chore i nie będzie nosicielem.
Ponieważ choroba może rozwinąć się tylko, jeśli oboje rodzice są nosicielami, w danej rodzinie może nie być znanej historii zachorowania. W połączeniu z bezobjawowym nosicielstwem tej mutacji, rodzice nie mogą być świadomi ryzyka związanego z posiadaniem potomstwa.
W chwili pojawienia się zachorowania należy wykonać testy u rodzeństwa.
W przypadku każdego kolejnego dziecka takiej pary istnieje 25% szansy na to, że odziedziczy obydwie mutacje, a tym samym będzie chorowało na MLD. Członkowie szerszego kręgu rodzinnego mogą się również przebadać na nosicielstwo mutacji ARSA. Szacuje się, że pojedyncza mutacja występuje u 1 na 100 osób.